DISEASES OF GLYCOLIPID METABOLISM
Glycolipids are a diverse class of bioactive molecules that function in a variety of normal and pathogenic cellular processes. Genetic defects in the enzymes that metabolize different glycolipids can lead to their abnormal accumulation of the lysosomes of different cell types. This accumulation can have a variety of pathogenic effects and lead to a collection of diseases called lysosomal storage diseases (LSD). Depending on the severity and the specific glycolipids involved, these LSDs can impact various body organs including the heart, kidney, liver and in some cases the central nervous system.
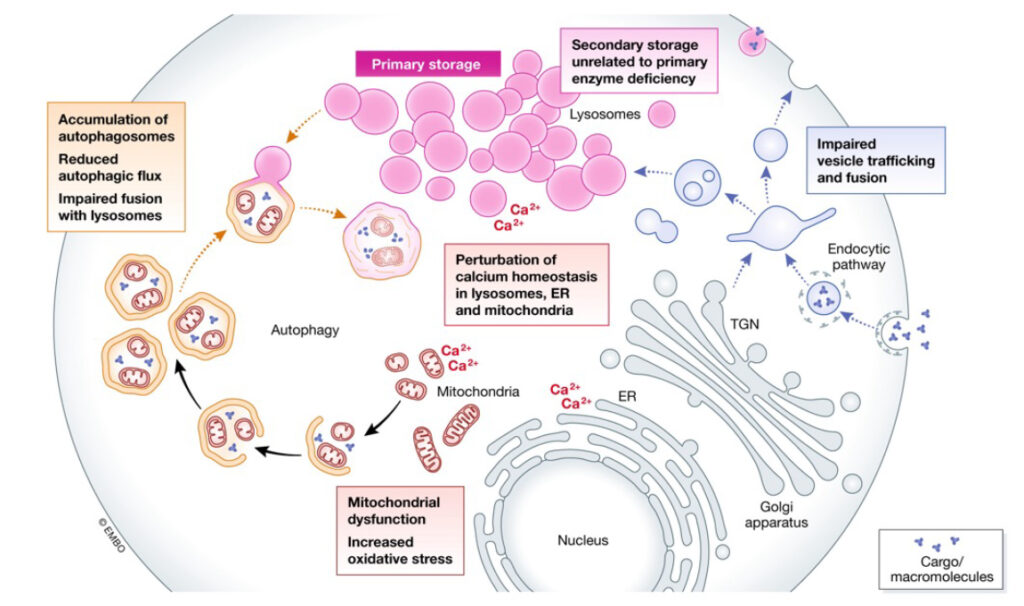 Parenti et al. EMBO MolMed. 2021
Parenti et al. EMBO MolMed. 2021
By inhibiting GCS, the key enzyme that synthesizes these glycolipids, we target the root cause of the diseases by decreasing the production of the pathogenic glycolipids. This approach is called substrate reduction therapy (SRT). AceLink is developing both brain-targeting and non-brain-targeting small molecule therapeutics to treat different lysosomal storage disorders. Some examples of LSDs that can be treated with SRT are Fabry disease, Gaucher disease, and GM1 and GM2 gangliosidosis.
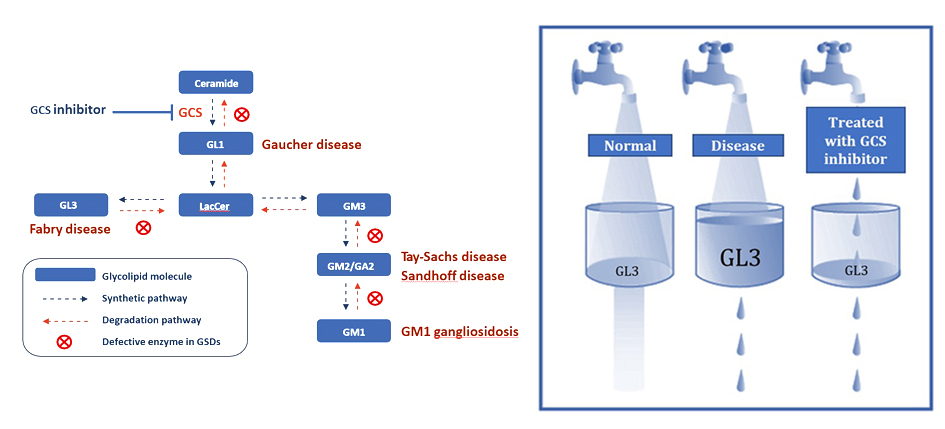
Glycolipids are also dysregulated in chronic diseases where they contribute to disease associated inflammation, cell proliferation, protein misfolding, and fibrosis. These diseases include polycystic kidney disease, diabetic kidney disease, fatty liver disease, heart disease, and even Parkinson’s disease. For these diseases, reducing the cellular levels of specific glycolipids can also be therapeutic.
FABRY DISEASE
GAUCHER DISEASE
GANGLIOSIDOSES
POLYCYSTIC KIDNEY DISEASE
GBA PARKINSONS' DISEASE
DIABETIC NEPHROPATHY (DN)
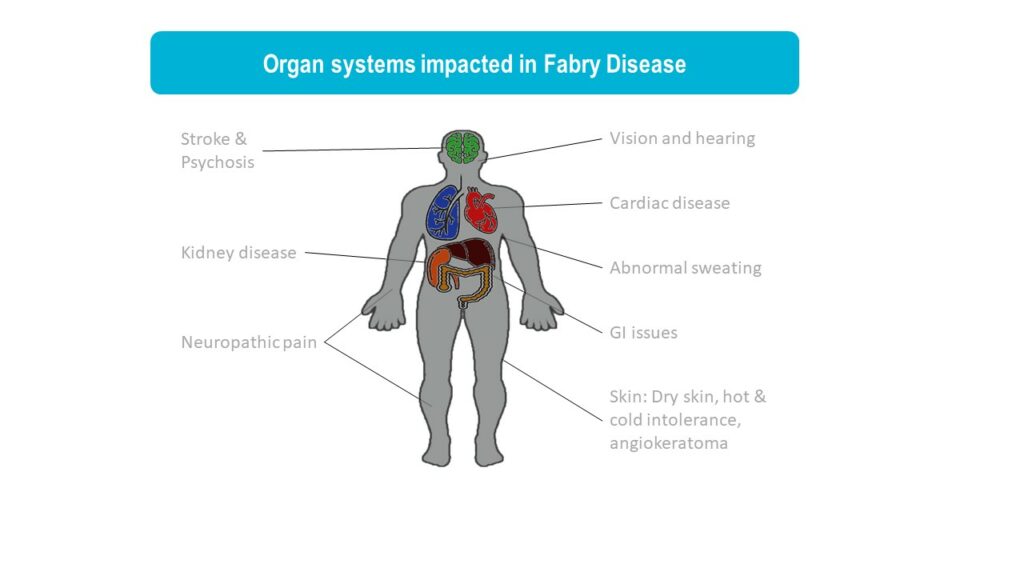
FABRY DISEASE
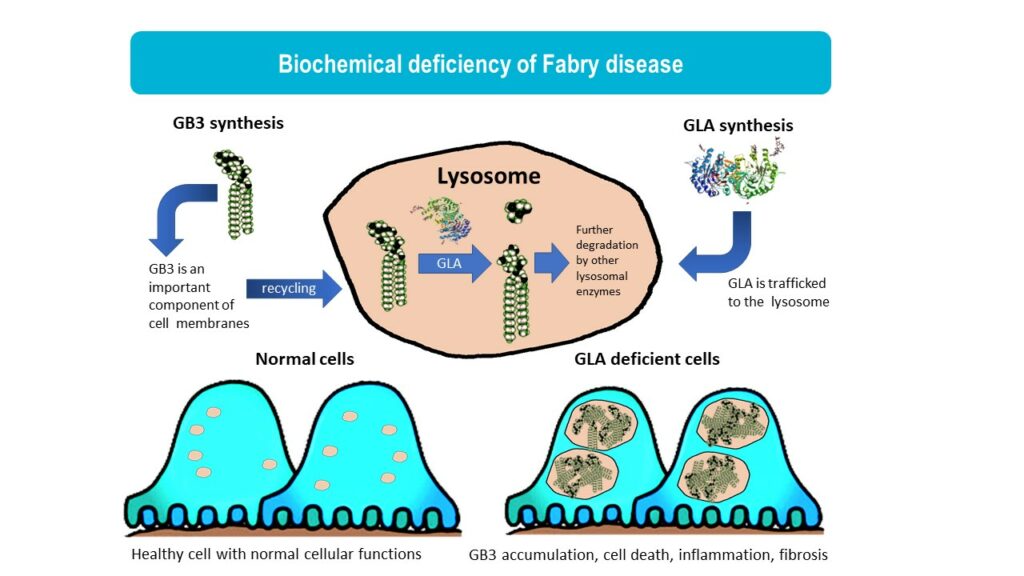
Fabry disease is a lysosomal storage disorder caused by mutations in the GLA gene, leading to reduced activity of the enzyme a-galactosidase A (αGal). In Fabry disease patients, αGal deficiency results in the accumulation of globotriaosylceramide (GL3), leading to a wide range of symptoms from pain, gastrointestinal issues, eye and skin problems to kidney failure, heart disease, and stroke. Fabry disease is an x-linked disorder and it is more commonly seen in males although females can also develop Fabry disease that is usually milder. Historically, it has been estimated that 1 in 50,000 males are affected by Fabry disease. However, with newborn screening, better awareness of Fabry disease in women, and identification of later onset forms of Fabry disease, it is now recognized that Fabry disease affects an estimated 1 in 1,000 to 9,000 people. (Medline)
Enzyme replacement therapy (ERT) is the standard of care for Fabry disease. While ERT reduces progression of many Fabry symptoms, the benefits are incomplete due to the low penetration of enzyme into some cell types. Many patients on ERT still develop progressive kidney and heart disease. There are other shortcomings of ERT, such as high cost, burden of life-long intravenous infusion, and allergic reaction to ERT. The limitations of ERT highlight the need for safe and effective small molecule therapies that are more efficient at penetrating diverse cell types and tissues.
AceLink is developing a GCS inhibitor AL1211 to treat Fabry disease.
GAUCHER DISEASE
Gaucher disease is a lysosomal storage disorder caused by mutations in the GBA1 gene. Mutations in GBA1 lead to reduced activity of the lysosomal enzyme glucocerebrosidase (GCase). In Gaucher patients, GCase deficiency results in the accumulation of glucosylceramide (GL1). Depending on the severity of the GCase deficiency, Gaucher patients can develop neurological symptoms as seen in type II and III Gaucher disease or they can develop milder non-neurological symptoms as seen in type I Gaucher disease.
Enzyme replacement therapy (ERT) and small molecule substrate reduction therapy (SRT) are available for Gaucher disease, but these therapies do not address the neurological features of Gaucher disease because they do not cross the blood brain barrier. AceLink is developing brain penetrant small molecule SRT that could be used to treat both CNS and non-CNS symptoms of Gaucher disease.
GANGLIOSIDOSES
GM1 Gangliosidosis is a lysosomal storage disorder caused by a mutation in the GLB1 gene. Mutation in GLB1 lead to reduced activity of the lysosomal enzyme beta-galactosidase (βGal). In GM1 gangliosidosis patients, βGal deficiency results in the accumulation of GM1 gangliosides (GM1). GM1 gangliosidosis patients with the most severe βGal deficiency develop severe early onset neurological symptoms while milder βGal deficiency can lead to later onset neurological symptoms.
GM2 Ganglisosidosis is a group of lysosomal storage disorders caused by mutations in the HEXA or HEXB genes. Tay-Sachs disease is a form of GM2 gangliosidosis caused by mutations in the HEXA gene and Sandhoff disease is caused by mutations in the HEXB gene. Together HEXA and HEXB encode protein subunits of the beta-hexosaminidase enzyme. Mutation in HEXA or HEXB lead to reduced activity of the lysosomal enzyme beta-hexosaminidase enzyme (βHex). In GM2 gangliosidosis patients, βHex deficiency results in the accumulation of GM2 gangliosides (GM2). GM2 gangliosidosis patients with the most severe βHex deficiency develop severe early onset neurological symptoms while milder βGal deficiency can lead to later neurological symptoms.
AceLink is developing brain penetrant small molecule SRT that could be used to treat GM1 and GM2 gangliosidosis.
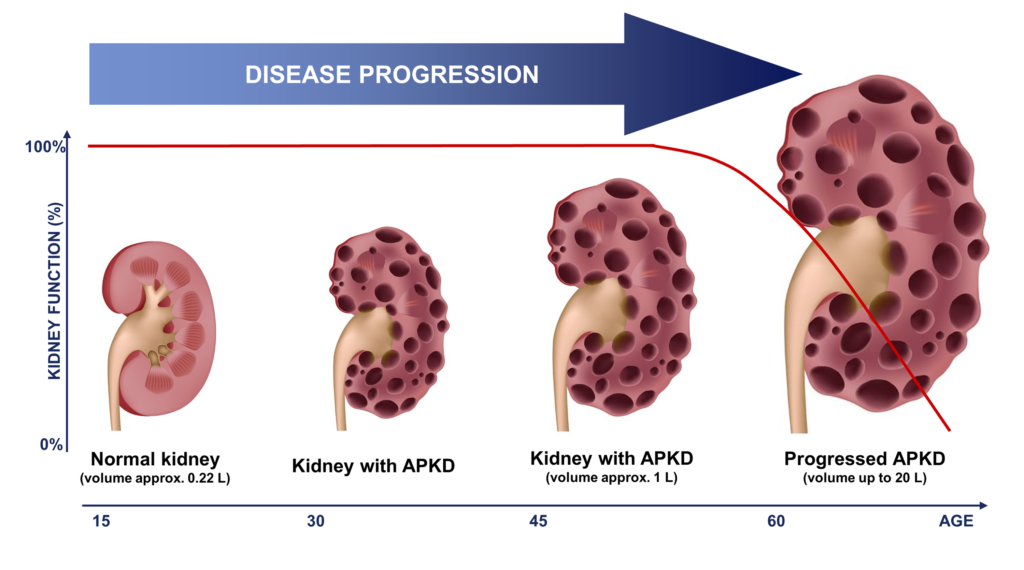
POLYCYSTIC KIDNEY DISEASE
Autosomal dominant polycystic kidney disease (ADPKD) is a genetic disorder in which numerous cysts develop primarily within the kidneys, causing the kidneys to enlarge and eventually lose function. ADPKD is the most common inherited kidney disease, characterized by the development of numerous fluid-filled cysts, kidney enlargement, and eventual progression to chronic kidney disease (CKD) and end-stage renal disease (ESRD). While some severe forms of ADPKD can affect young children, most PKD patients develop symptoms in early adulthood or later. The probability of progressing to ESRD increases with age, with 50% of ADPKD patients over the age of 60 years of age requiring dialysis or kidney transplantation. The majority of ADPKD cases are caused by a mutation in one of two genes, PKD1 or PKD2. These genes encode two proteins that localize to the primary cilia of renal epithelial cells where they play an important role in regulating renal development and function, promoting epithelial differentiation, and preventing cell proliferation.
The glycosphingolipids GL1 and GM3 are elevated in kidney tissue from patients with polycystic kidney disease and in animal models of polycystic kidney disease. Studies using GCS inhibitors in multiple different animal models of PKD have demonstrated that reducing the production of GSLs can slow cyst growth and preserve kidney function.
Natoli TA, Modur V, Ibraghimov-Beskrovnaya O. Glycosphingolipid metabolism and polycystic kidney disease. Cell Signal. 2020 May;69:109526. doi: 10.1016/j.cellsig.2020.109526. Epub 2020 Jan 10. PMID: 31911181.
Natoli TA, Smith LA, Rogers KA, Wang B, Komarnitsky S, Budman Y, Belenky A, Bukanov NO, Dackowski WR, Husson H, Russo RJ, Shayman JA, Ledbetter SR, Leonard JP, Ibraghimov-Beskrovnaya O. Inhibition of glucosylceramide accumulation results in effective blockade of polycystic kidney disease in mouse models. Nat Med. 2010 Jul;16(7):788-92. doi: 10.1038/nm.2171.
GBA PARKINSONS' DISEASE
GBA Parkinson’s disease is arguably the most common genetic form of Parkinson’s disease. Numerous genetic studies have shown that a single mutation in the GBA1 gene (the gene that causes Gaucher disease) can increase your risk of developing Parkinson’s disease by up to 30-fold. In vitro and in vivo preclinical studies suggest that the protein alpha-synuclein can interact with glycolipids and this can lead to aggregation and toxicity. Studies using GCS inhibitors in mouse models of Parkinson’s disease support that lowering the production of these glycolipids can reduce synuclein aggregation, slow the progression of neurobehavioral phenotypes, and neurodegeneration.
Vieira SRL, Schapira AHV. Glucocerebrosidase mutations and Parkinson disease. J Neural Transm (Vienna). 2022 Sep;129(9):1105-1117. doi: 10.1007/s00702-022-02531-3. Epub 2022 Aug 6.
Sardi SP, Viel C, Clarke J, Treleaven CM, Richards AM, Park H, Olszewski MA, Dodge JC, Marshall J, Makino E, Wang B, Sidman RL, Cheng SH, Shihabuddin LS. Glucosylceramide synthase inhibition alleviates aberrations in synucleinopathy models. Proc Natl Acad Sci U S A. 2017 Mar 7;114(10):2699-2704. doi: 10.1073/pnas.1616152114. Epub 2017 Feb 21.
DIABETIC NEPHROPATHY (DN)
Diabetic nephropathy (DN) is a common form of kidney damage that occurs with the progression of metabolic diseases like type I and type II diabetes. If diabetes is not properly treated then damage can occur to the glomeruli, the filtration units of the kidney. This damage can lead to high blood pressure, which puts more pressure on the kidneys and leads to more damage, chronic kidney disease, (CKD) and eventual progression to end stage kidney disease (ESRD).
Glycolipids have been shown to play roles in metabolic disease. Elevated levels of glycolipids contribute to insulin resistance and lowering these glycolipids with GCS inhibitors can increase insulin sensitivity and improve glucose tolerance. Glycolipids are elevated in the kidneys of diabetic nephropathy patients where they likely contribute to inflammation, hypertrophy, and fibrosis. Therefore, treating patients with GCS inhibitors can target both the metabolic disease and the resulting tissue damage occurring in DN.